
Article
Why Ecosystem Design Controls Are Really About Moving Faster With the Right Rigor

Software companies in the digital/connected era are using the “everything’s connected in real time” digital infrastructure to build and continuously improve robust products at ever increasing speed.
These same methods and the infrastructure that supports it hold tremendous promise for the healthcare sector including medical device, diagnostic and pharma companies currently regulated by the FDA.
The FDA has recognized that their “traditional approach to moderate and higher risk hardware-based medical devices is not well suited for the faster iterative design, development, and type of validation used for software-based medical technologies.”
How can they adapt so they fulfill their mission of ensuring patient safety and efficacy while not standing in the way of better and faster innovation? And how will that impact medical device and digital health companies?
Over the last few months there have been a couple of interrelated developments at the FDA that address this question, and will likely have a big (and mostly beneficial) impact on companies incorporating digital health into their product offerings.
In May, Bakul Patel, the FDA’s associate director of digital health, announced that the department would be hiring 13 additional staff to form a digital health unit focused on Software as a Medical Device (SaMD), software inside medical devices (SiMD), interoperable devices and other novel digital health technology. He provided more context in a talk at Xavier MedCon in Cincinnati, and in interviews for articles in Wired and other publications.
On June 15th, Scott Gottlieb, the new FDA Director, wrote an article in FDA Voice entitled Fostering Medical Innovation: A Plan for Digital Health Devices. that outlined a new digital health innovation plan
On July 27th, Gottlieb announced a new pilot program for pre-certification and published a new Digital Health Action Plan outlining future tasks and goals.
Finally, on August 1st, Patel held an informational webinar about the pre-certification pilot program for digital health companies.
1. A Digital Health Unit with 13 New Hires.
The new group is being formed and new staff is being hired to better address the rapid innovation in digital technologies that are applicable across many clinical areas, and to accomplish the other tasks outlined in the Digital Health Action Plan. Topics they’ll focus on will include artificial intelligence, advanced analytics, the cloud, wireless medical devices, telemedicine, interoperability, health IT and cybersecurity.
Those extra staff will definitely be necessary to handle the tasks in the digital health action plan
2. More Clarity on Scope of Oversight and Approach to Digital Technology
• Guidance on what falls out of scope and what’s low enough risk to not subject them to pre-market regulatory requirements. This will be a continuation of the efforts the FDA has been making to narrowly define and further clarify regulatory scope. To quote Fostering Medical Innovation: “Greater certainty regarding what types of digital health technology is subject to regulation and regarding FDA’s compliance policies will not only help foster innovation, but also will help the agency to devote more resources to higher risk priorities.”
• Reduce ambiguity on the FDA’s approach to new technology
As Gottlieb wrote, on June 15th (link), the FDA is looking to create “policies that are clear enough for developers to apply them on their own, without having to seek out, on a case-by-case basis, the FDA’s position on every individual technological change or iterative software development..”
This is particularly important for things like cloud computing (what constitutes a controlled environment?), machine learning (how do you validate and test?) and complex systems with multiple sub systems, including regulated and unregulated components. The latter, dubbed Mulitfunctionality, will be specifically addressed in an upcoming draft guidance.
3. Development of a more efficient, risk-based regulatory framework for overseeing digital health technologies.
This is starting on September 1st with the pilot program for pre-certification of companies, rather than products. That will enable pre-certified companies to “market and iterate on products without continuing to knock on the FDA’s door, or through streamlined pre-market review.”
Pre-certified companies would need to submit less information, or in some cases no pre market submission at all. In that case, they could launch a new product and immediately begin post-market data collection. demonstrate that the underlying software and internal processes are sufficiently reliable. Post-market data could help FDA assure that the new product remains safe and effective as well as support new users.”
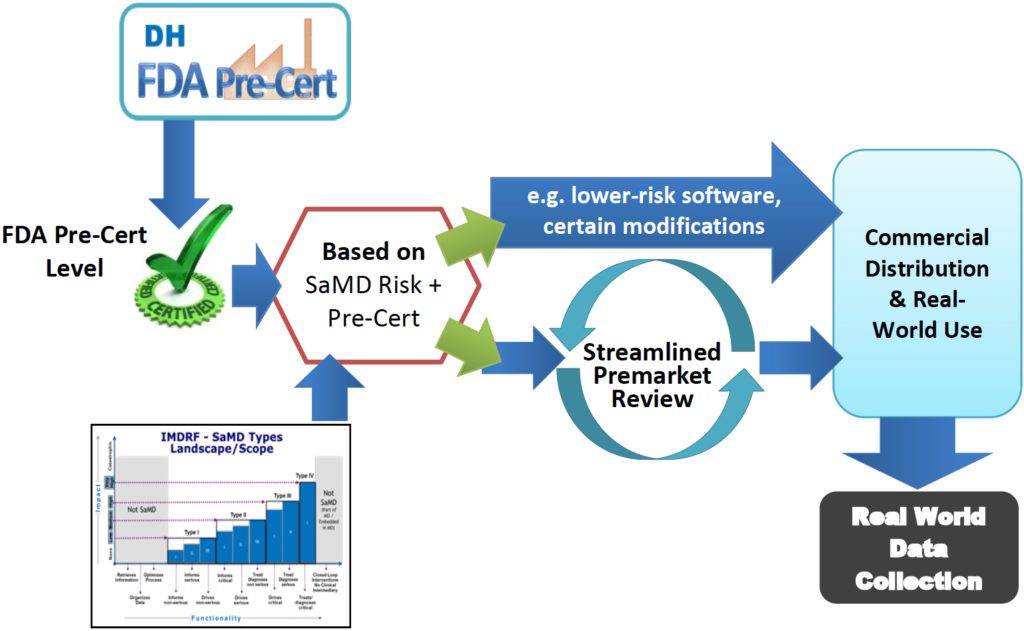
4. Using post-market collection of real world data.
This has been one of the key drivers of connected age product improvement in other industries, and is something the FDA intends to use for both regulatory oversight and fostering innovation.
Pre-certified firms could “collect real-world data postmarket that might be used, for example, to affirm the regulatory status of the product, as well as to support new and evolving product functions.”
The FDA could also use better post market surveilance data, either gathered by firms themselves, or through groups like the National Evaluation System for health Technology (NEST). NEST will be a federated virtual system for evidence generation composed of strategic alliances among data sources including registries, electronic health records, payer claims, and other sources.
While the scope of the initial pilot project is limited to Software as a Medical Device (SaMD) products, the FDA hopes the knowledge is transferrable to other areas. The FDA will continue to emphasize better collection and evaluation of post-market real world evidence. And companion software for medical devices, diagnostics and pharma will continue to be more deeply interconnected, and more integral to those products value propositions.
Companies that develop expertise in and best practices for applying “build-measure-learn” software practices within the constraints of FDA quality systems regulations will be able to move faster to take advantage of market and policy changes focusing on real-world outcomes and value-based care.
We’ve covered some of these best practices in our ebook on Agile in an FDA Regulated Environment, and we’ll be be addressing others, like testing automation, user analytics, and testing harnesses on this blog in the coming weeks.
Related Posts
Article
Why Ecosystem Design Controls Are Really About Moving Faster With the Right Rigor

Article
MedTech Teams Should Stop Paying for Compliance Twice

Article
Beyond the Device: Where MedTech Digital Ecosystems Are Starting to Pay Off

Article
Why MedTech Teams Slow Down as Software Grows and What Better Architecture Looks Like
