
Article
Roundtable Chapter 2 – The One With the Bottleneck at the Finish Line

Recent years, we have seen digital health technologies moving toward rapid adoption in the United States. Yet, Software as a Medical Device (SaMD), mobile medical devices, digital therapeutics (DTx), and Device manufacturers with artificial intelligence (AI) components are often anxious about the lengthy and detailed process of the existing FDA’s premarket review for medical devices. The FDA Software Precertification (Pre-Cert) Program, announced in August 2017, aims to streamline the current premarket review process and reduce unnecessary regulatory burden for both medical device software manufacturers and the FDA. The Pre-Cert program can be thought of as the FDA’s response to an increasingly complex, evolving ecosystem of digital health, as well as the fast-paced technological innovation that has been driving change across virtually every sector of the economy. The program’s primary goals — to develop effective medical device software, drive faster innovation, and enable timely patient access, all while keeping safety at the forefront — are part of the FDA’s continuing efforts to maximize the effectiveness of their processes around the development and regulation of medical device software.
The Pre-Cert Program is just one of several large initiatives the FDA has undertaken in the last few decades to better adapt their regulatory processes to the changing technological landscape. For example, In a companion article, we take an in-depth look at Software as a Medical Device (SaMD) — what it is, what it does, and how healthcare companies can take advantage of this special category of medical device software.
In this article, we explore the Pre-Cert program in the context of the FDA’s overall strategy, look at how it started back in 2017, assess progress after 4 years of testing the program, and provide medical device companies with a working guide on how to leverage these regulations to prepare for what lies ahead.

Portions of Title 21 of the Federal Code of Regulations, which govern the FDA’s oversight of medical devices, were put in place after the 1976 Medical Device Amendment of the 1938 Federal Food, Drug and Cosmetics Act. While these regulations have been amended a number of times since 1976, the core has been the same for over 40 years.
At the time, there was not a lot of software in medical devices (and very little in the world in general).
However, since then, there have been a series of innovations that have served to accelerate the prevalence and importance of software in our daily lives and in every sector of the global economy, including personal computing, the internet, mass-market cell phones, smartphones, and the internet of things.
Medical devices are no exception, and software now plays a vital role in creating opportunities to improve the patient experience, improve health outcomes, reduce the per capita cost of care and drive faster innovation.
The pace of innovation for software is much faster than for traditional medical devices. This is especially true when it comes to Software as a Medical Device (SaMD), which can function as an autonomous medical device without the constraints of slower hardware development, production and distribution.
Because of the increasing importance of software and the ever-quickening pace of software-driven innovation, the FDA recognized the need to move away from an outdated regulatory model and move towards a model that prioritizes faster innovation, without sacrificing patient safety.
The FDA’s move towards a different regulatory model was also, in part, prompted by Congress’s 21st Century Cures Act, “designed to help accelerate medical product development and bring new innovations and advances to patients who need them faster and more efficiently.”
As a result, one of the FDA’s primary initiatives over the last few years has been the Digital Health Innovation Action Plan, which, among other things, has seen the FDA work to develop a regulatory framework around SaMD that mitigates risk without hindering progress, also known as the Software Precertification (Pre-Cert) Program.
The FDA’s latest regulatory framework, 2017’s Digital Health Software Precertification Program, builds on its previous programs, including the Case for Quality program (discussed later), and tailors it to medical device software, including SaMD.
According to the FDA, software in medicine continues to increase in popularity, thanks in part to its ability to “treat and diagnose conditions and diseases, aid clinical decision making, and manage patient care.”
In other words, SaMD has the potential to improve health outcomes while leveraging increased connectivity (the internet, smartphones, the cloud), data, and the “internet of everything” to do more, cheaper and faster.
At the time, however, the FDA lacked the framework needed to effectively drive this type of innovation. Its Software Precertification Program was designed to enable:
Compared to traditional medical devices with the constraints of slower hardware development, production and distribution, SaMD products can take advantage of faster development, testing, real-world performance data collection, and updates or improvements to the product.

Earlier, we highlighted how the Software Precertification Program is part of the FDA’s Digital Health Innovation Action Plan (DHIAP), which seeks to refine and improve the process by which digital health innovations are reviewed by the federal agency.
The ultimate goal of this plan is to provide timely access to high-quality, safe, and effective medical technologies utilizing the latest advances, while driving innovation under accordance of the 21st Century Cures Act.
Before we address some of the goals of the Pre-Cert Program, we want to highlight two important changes under the DHIAP:
1) The expansion of the FDA’s Digital Health Unit
Published in July of 2017, the Digital Health Innovation Action Plan outlines future tasks and goals to address rapid innovation in digital health technologies.
Among these announcements was the establishment of a Digital Health Unit (including 13 new hires), which deals with complex 21st century issues such as artificial intelligence, advanced data analytics, cloud computing, wireless medical devices, telemedicine, interoperability, health information technology, and cybersecurity.
A key benefit of this new Unit includes the alignment of the device review process with the software development life cycle, which will speed up patient access to potentially lifesaving technologies and improve transparency for both the manufacturers and end users of these products.
2) More clarity on the scope of oversight and approach to digital technology
The DHIAP program offers better guidance on the FDA’s regulatory scope of digital health technologies, which also serves to eliminate ambiguity concerning the agency’s approach for the developers of such technologies.
This is particularly important for high-tech software concepts including, but not limited to, cloud computing, machine learning, and complex systems, which remain in early stages of development.
The expected result is a fostering of faster innovation, as well as better resource allocation within the agency towards high-risk priorities.
Goals of the Software Precertification Program, which fall under the DHIAP and which will also serve as a test for broader adoption, include:
1) The development of a more efficient, risk-based regulatory framework for overseeing digital health technologies
The FDA cannot use their old regulatory paradigm to deal with SaMDs because they will be overwhelmed by the volume of work. Such a backlog will ultimately constrain and slow innovation. The Pre-Cert Program ensures that the regulatory approval process is more timely and relevant for software devices.
Pre-certified companies will experience a streamlined premarket review process, where they can submit reduced documentation in order to bring their product to market. As a result, these companies will be able to launch SaMD products much faster and immediately begin the post-market data collection phase to ensure the safety and efficacy of the product.
If the FDA pilot is successful and they can get large scale adoption on SaMD, they will likely seek to extend this updated framework beyond De Novo SaMD into 510(k)s and into connected devices with extensive SiMD. This will rapidly accelerate the pace of innovation and discovery in the medical field.
2) More post-market collection of real-world data
Post-market data is required by the FDA in order to assess the safety and effectiveness of an FDA-approved device. A main advantage of the Pre-Cert Program is that it emphasizes post-market collection of real-world data.
Because SaMD (as well as SiMD) often utilize sensors, software and internet connectivity, post-market data can be acquired more easily and more cost effectively, potentially enabling a faster “build-measure-learn” cycle to speed up innovation.
Both manufacturers and the FDA can then use this data to improve product safety and efficacy, as sharing post-market data with the FDA enables better oversight.
3) Enable faster patient access to technologies
Software development can be done faster than traditional hardware development; you can get faster feedback to improve your product, and you can distribute those changes to the marketplace much more quickly.
In short, the FDA’s old paradigm was simply too slow.
To support its drive for faster innovation, the FDA realized that certain critical components of the software development process had more to do with a company and its processes than a particular product.
As a result, the FDA has shifted its focus to certify companies and their core processes, while relying on them to effectively execute such processes. This frees the FDA to focus on actual product-specific clinical validations for product oversight, rather than spending its time analyzing whether or not a company is following the correct product development process.
(This is analogous to the Case for Quality pilot and ISO 13485 certification, which are also at a company level and discussed below.)
In short, the precertification program ensures that a company and its processes are, as its name implies, pre-certified to conduct this important work. Pre-certified companies will benefit from a streamlined approval process to get their products into the marketplace, faster.

In the middle of 2017, the FDA selected nine companies to participate in the first-of-its-kind pilot program, known as the Software Pre-Cert Pilot Program, designed to change how digital health is regulated in the US.
Making an appearance on the list are some familiar names, including top players in tech (Apple, Google, Samsung and FitBit) and well-known medical device and pharmaceutical companies (Johnson & Johnson, Roche).
The list also includes smaller and emerging tech companies, such as Pear Therapeutics, a digital therapeutics company and the not-for-profit startup, Tidepool, which develops free software for the diabetes community.
This broad range of companies both large and small — from well-known multiline medical device and pharmaceutical companies to smaller digital therapeutics startups — was intentional. The FDA wanted to make sure different perspectives were taken into consideration, and that the working model/precertification program would not put small companies at a disadvantage.
The FDA will base precertification around five excellence principles, which remain in development but currently include:
The precertification program’s excellence principles and evaluation criteria seek to incorporate best practices for developing software products.
In fact, one of the main goals is to integrate those best practices other companies have already learned in order to build the best possible SaMD products — products that support an increasingly connected world, yet keep the high standards of patient safety and product quality required in healthcare at the forefront.
Following the FDA’s excellence principles is critical to success in healthcare and beyond. The principles not only act to guide companies towards precertification, but serve as a roadmap of best practices regardless of whether a company is seeking FDA approval for their product or not.
Any company wanting to build a great product for today’s connected world should follow these five principles. Why? Because as touched on above, they have already been synthesized from the best practices outside of the medical device industry, and then modified to ensure that clinical effectiveness and patient safety remain top priority.
With the Pre-Cert Program, the FDA has not only confirmed that the best practices developed in other industries are valid, they are explicitly stating that they will reward companies who follow them. And for a generally risk averse industry, the fact that the FDA has given its stamp of approval shows just how valuable these principles are.
In short, there are nothing but positive outcomes for companies that follow the five excellence principles. The same principles apply to both Software in a Medical Device (SiMD) and SaMD, and all companies in healthcare-related industries should understand how to apply these principles to their products to gain a competitive edge in the marketplace.

As we’ve seen, the FDA recognizes the pressing need to change its regulatory paradigm in the face of accelerating innovation in today’s “internet of everything” world.
That need is not going away.
But what about the FDA’s proposed solution to the problem, the Pre-Cert Program? Will this model stand the test of time?
We believe that it will, for two main reasons:
First, as discussed above, the Precertification program actually aims to solve a major problem plaguing both the FDA and the industry as a whole: how to ensure better safety and efficacy, while lowering the burden of regulatory oversight;
Second, the Pre-Cert Program is not an outlier to the FDA’s strategy, but rather a part of a larger, more coherent strategy.
The Pre-Cert Program rests on two main pillars:
These same pillars are not isolated within the Pre-Cert Program, and are reflected in the FDA’s other initiatives, including:
Let’s explore each in greater detail.
Nearly a decade ago, the FDA recognized that their paradigm of looking at each and every medical device independently of who was building it, while also evaluating pre- and post-market data separately, was insufficient in a world where the fast feedback loop of “build-measure-learn” software innovation was quickly becoming the norm.
Since then, they have been working to create and clarify a regulatory framework for medical devices and medical device software in a way that prioritizes both patient safety and healthcare innovation.
In 2011, the FDA undertook a review of medical device quality data and feedback from FDA officials and industry stakeholders. During this review process, the FDA identified several common manufacturing risks that were impacting the quality of medical devices.
Their analysis revealed that when manufacturers took steps to mitigate these risks, they saw benefits such as higher productivity, fewer complaints, and lower quality-related costs. With evidence that investments in quality have significant long-term returns, the FDA launched the Case for Quality program.
The Case for Quality program helps the FDA identify and distill best practices that enable certain device manufacturers to consistently produce high-quality devices. It allows them to promote such practices and focus resources on supporting other manufacturers in raising their quality standards.
The program also provides a framework for the FDA to work with industry stakeholders to align regulatory, enforcement and compliance efforts with best practices.
Included in the Case for Quality program are pilot programs intended to drive quality and evaluate the current regulatory environment. Launched in early 2018, the Voluntary Medical Device Manufacturing and Product Quality Pilot Program removes regular FDA surveillance and pre-approval inspections for a select group of manufacturers, opting instead for periodic appraisals by certified third-party teams.
The goal is to drive “continuous improvement and organizational excellence among participating medical device manufacturing sites” through these quality appraisals and reduce the burden and disruption that manufacturers typically face when undergoing FDA inspections.
Similarly, the Premarket Approval (PMA) Critical-to-Quality Pilot Program enables qualified PMA device manufacturers to streamline the preapproval process by foregoing a preapproval inspection and instead proactively engaging with the FDA early in the PMA review process to assure that critical quality criteria are present.
In early 2018, the FDA announced it would be aligning U.S. medical device regulations from FDA 21 CFR Part 820 (which defines the minimum criteria medical device companies need to meet to sell their products in the United States) to ISO 13485:2016, which evaluates a company’s quality management systems, and serves as the standard recognized by much of the rest of the world’s medical device market.
Just as the Pre-Cert Program envisions companies being pre-certified, there is a certification process for ISO 13485, administered by notified bodies such as Tüv Süd, BSI, Intertek, etc. in which a company’s quality management system(s) is periodically certified and audited to the standard.
Late in 2018, the FDA officially announced an ambitious reorganization of their pre-market and post-market review process to address total product life cycle (TPLC). This new restructuring, under the Office of Product Evaluation and Quality (OPEQ), serves “to create an agile infrastructure that can adapt to future organization, regulatory and scientific needs.”
Additionally, “this new approach is expected to help facilitate information sharing to allow for more informed decisions, ensure process and policy consistency, and provide more straightforward and streamlined interactions.”
In fact, both the Case for Quality Program and the Software Precertification Program are part of the FDA’s larger shift towards a total product lifecycle approach. The significance of this is that the FDA isn’t just taking into consideration the digital “maturity” of the companies it’s evaluating, but is actually changing its own organization (the FDA itself) to reflect this.
It’s clear that the FDA has placed significant importance on transforming software regulation across the medical device space. In addition to its Pre-Cert Program and aforementioned initiatives, the FDA has provided additional guidance across several key areas related to the Digital Health Action Plan. These include:
While nothing is certain just yet, we can see a trend emerging where the precertification program model is being woven into the agency’s thinking around the regulation of software as a whole, and we expect that this will continue to impact how the FDA operates moving forward.
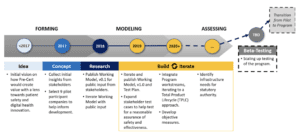
In early 2019, the FDA held a webcast where it reviewed milestones of the 2017 Pre-Cert Pilot Program and released the next phase of the pilot program, dubbed the Pre-Cert 1.0 and also released the 2019 Test Plan. This iteration of the pilot will be limited to a select number of manufacturers of SaMD.
For these participating organizations submitting a premarket application, the FDA will apply both the Pre-Cert model (streamlined premarket review and excellence appraisal) and the traditional review process to demonstrate that the Pre-Cert model can still provide the same quality of data for evaluating whether a product meets the FDA’s safety and effectiveness standards. The results from this pilot phase are intended to inform a future Pre-Cert framework.
The FDA has made it clear they recognize the need to adapt to new advances in technology and the speed of innovation to help transform how care is delivered. The Software Pre-Cert Pilot Program, along with the shift towards a total product life cycle focus, have laid a foundation for the FDA to speed up innovation, without losing sight of patient safety.
It’s crucial for manufacturers and developers to understand the different regulations applicable to their products. Receiving FDA precertification will not only help accelerate the proliferation of digital health software but will act as a springboard for entry into the marketplace.
In September 2020, the FDA updated the Digital Health Software Precertification (Pre-cert) Program website with a new summary document summarizing learnings and ongoing activities during the build and iterate phase. The report highlighted key takeaways from the initial pilot testing with the nine pilot companies and the next steps for the future of the Pre-cert Program.
The September 2020 report discussed how future phases of the Pre-cert Program will include refinements across the program to drive repeatability of the processes, improve the quality and quantity of information, provide clarity and reduce time burden on both internal and external stakeholders. The following are notable action items defined to make progress on the Pre-cert Program:
The healthcare industry is closely following the FDA Software Pre-Cert Program for regulation of Software as a Medical Device (SaMD) and Digital Therapeutics (DTx) products. The program aligns with the TQM approach with a focus on everyone working together to continuously improve the overall process. We believe that the Precertification Program has great potential for advancing the development of safe and effective digital health products.
As of this writing, it is still unclear when the FDA will move forward with their beta testing and officially launch the program according to Figure 1: Software Pre-Cert Pilot Program Development Roadmap below. There are no proposed dates for next phases of the program. The Pre-Cert Pilot Program relies heavily on collaboration to develop an effective model for SaMD developers. Along with the summary, the FDA also released an updated Frequently Asked Questions (FAQs) on the Pre-Cert Program to provide more insight into the Pilot. You can follow the updates to the Pre-cert Program here.
Unsurprisingly, given the ambitious nature of this program, there are a variety of opinions among industry experts about the successes, challenges, and opportunities facing the Pre-Cert program. While Orthogonal does not necessarily endorse any one opinion or another, we are providing the following list of readings if you want to further delve into the rich conversation around Pre-Cert.
“Establishing a Regulatory Framework for Digital Health Medical Devices Regulatory pathways under current authority do not promote flexibility for FDA to optimally regulate emerging technologies. This is exemplified in the digital health space, where the current statutory framework for regulation of medical devices is not well suited to the faster cycle of innovation, iterative design and development, and validation approaches used for digital health devices. FDA proposes establishing a framework that would allow FDA to tailor and apply the appropriate requirements for the reasonable assurance of safety and effectiveness for a software as a medical device product based on the risk of the product and throughout the product lifecycle.”
In a future article, we will discuss more about how the FDA will be leveraging the organization-based total product lifecycle (TPLC) approach from the Pre-Cert Program to build upon the regulation of AI in a recent FDA “Proposed Regulatory Framework for Modifications to Artificial Intelligence/ Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) – Action Plan” document published on January 2021. We would not be surprised if this AI/ML SaMD draft guidance scheduled to come out in late 2021 were to be heavily influenced by the position paper and thinking around the Pre-cert Working Model.
Globally, the FDA is leading the way with its digital health regulatory efforts. In fact, the agency plays a significant role in the International Medical Device Regulators Forum (IMDRF), where regulators from around the world work to advance and harmonize standards.
Increasingly, SaMD is at the forefront of their conversations.
While other countries have also developed regulations around SaMD (as well as medical software in general), the FDA’s initiatives place them far ahead of the rest of the international community. Still, several countries are making notable strides. These include:
When it comes to SaMD, the IMDRF framework plays a key role in pushing for a common language and global alignment around certain quality management standards while respecting local regulatory considerations.

As we have seen, the Pre-Cert Program is necessitated by the FDA’s need to deal with the continually increasing importance of software — and with the fast-paced change that occurs within software — coupled with its desire to take advantage of modern best practices to improve patient safety and foster faster innovation.
Such a move is not new, and fits within the FDA’s overall TPLC approach towards the regulation of medical devices.
Both the lessons learned, along with various aspects of this new paradigm, will permeate into any area of medical devices where software is becoming more important — which is to say, almost every aspect of medical devices.
The good news is that the excellence principles and metrics the FDA is looking at are highly compatible with best practices that have already proven effective across other industries for success in the IoT era.
One way to think about this is that the FDA is giving companies permission to apply these best practices. Remember: this is an industry that seems to require FDA permission for everything, so this is a big deal.
As in other industries, companies that embrace these principles, regardless of whether they are doing SaMD or SiMD, are more likely to succeed.
It’s in everyone’s best interest to integrate FDA best practices into the development of SaMD, as it will lead to quicker innovation, safer products, and faster turnaround with the FDA. The FDA’s five excellence principles lay the foundation and provide the guidelines for fostering innovation and clinical safety while staying competitive within the marketplace.
If you are a company that is producing SaMD, should you look at joining and piloting in the FDA’s Software Pre-Cert Program? This will depend largely on where you are in your product life cycle.
If you are working on your first SaMD product and cannot yet think of yourself as a company with a platform for SaMD products, or have a portfolio of SaMD products, this is probably not the right route for you (yet). However, if you are a company with a platform and a pipeline of products at different stages, the Pre-Cert Program could be advantageous for streamlining regulatory oversight.
Even if you are not yet ready to join the Pre-Cert Program, you should still pay attention to developments in this space and follow the FDA’s five excellence principles. These best practices will help you to innovate while maintaining clinical effectiveness and patient safety and to better compete in the marketplace.
For those companies developing SiMD but not SaMD, the same forces of our connected world still apply. This means that the evolution of the FDA regulatory environment and the best practices for developing SaMD are just as relevant. The Pre-Cert Program is focused on SaMD right now because the FDA had to start somewhere and keep the pilot small enough to evaluate and refine, but its learnings and approach have broader implications that we should eventually see applied to SiMD.
This is part of a wider trend as the FDA reevaluates its regulatory framework in favor of an approach that can keep up with today’s “internet of everything” world while fostering innovation and protecting patient safety. The FDA is committed to moving in this direction, and thus the pre-cert program should be carefully watched by companies operating in this space.
The FDA recognizes the enormous potential of digital innovation in healthcare, while being acutely aware of the regulatory, cost and patient safety challenges the industry faces. The past decade has seen the agency make substantial strides in its effort to lead the rest of the world in regulatory reform that responds to the reality of how medical device software is developed, while promoting best practices throughout the industry.
Over the past few years especially, the FDA’s efforts have accelerated, and the agency is moving quickly to test a broader framework that will promote innovation while still protecting patient safety.
The FDA has been focusing on how companies, as well as the FDA itself, can apply a total product lifecycle approach to drive faster innovation, and, through its five excellence principles, have provided both explicit direction and a direct path to do so.
Moreover, the healthcare industry is aligned with the FDA’s efforts, making it in every company’s best interest to incorporate these principles into their product development lifecycle.
While today’s connected “internet of everything” world is particularly apparent in the Software as a Medical Device space, where innovation is happening at a rapid pace, it will have a wider reach to other healthcare-related industries, including pharmaceuticals.
Thus far, the medical device industry, tech giants, pharma, leading multi-line healthcare companies, as well as nimble startups, have all been receptive to these changes, and have been active participants in the review processes and pilot testing of new initiatives.
Regulatory change in healthcare and medical devices is moving quickly and fits within the larger scope of digital innovation within the industry. While nothing is certain, these efforts align with where the industry– and the wider world –is headed, and we expect these trends to continue through the SaMD space and beyond.
Orthogonal integrates modern agile product development best practices with patient safety and regulatory compliance to build SaMD and connected medical device systems. If you need help building your next SaMD, or to learn more, contact us or call us at (312) 372-1058.
Related Posts
Article
Roundtable Chapter 2 – The One With the Bottleneck at the Finish Line
Article
Roundtable Chapter 1 – The One Where the Device Was Only Half the Story
Article
Roundtable Prologue – The One Where the Algorithm Couldn’t Sell Itself

Article
Managing Emerging Risks in SaMD: Strategies for 2025 and Beyond
