
White Paper

This blog contains Part 1 of the Orthogonal and MedSec white paper titled “Making the Significant Insignificant: Implementing a Predetermined Change Control Plan (PCCP) for Class II SaMD Products Beyond AI/ML.” The following are links to each part of this white paper:
The continued growth of Software as a Medical Device (SaMD) means that a large number of people are currently engaged in SaMD products. But there’s a smaller group of people who have successfully developed SaMD and gotten it to market. This minority of “been there done that” people have valuable lessons to share that have heretofore gone untapped.
Orthogonal and MedSec saw the opportunity to bring together these experienced people from a variety of companies to tackle tough SaMD questions. Last winter, we met in Florida for two days to work through a problem facing our entire industry – how to widen the umbrella of what falls under Predefined Change Control Plans, now that the 2023 Congressional Omnibus Bill calls for the FDA to support it.
Our group discussed how to expand that umbrella in a responsible and effective way. We took a look at the Congressional language to evaluate software solutions beyond AI/Machine Learning that could benefit from a PCCP. We excitedly offer up our results to our peers in SaMD development and the FDA, with the hope that it’ll help all of us move forward.
– – – – –
The U.S. FDA first referenced the term “Predetermined Change Control Plan” (or PCCP) in its April 2019 discussion paper[1] titled “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) – Discussion Paper and Request for Feedback” (referred to thereafter as 2019 FDA PCCP Discussion Paper). The goal was to develop a new regulatory framework to manage devices using AI/ML technologies, including continuously learning or adaptive AI/ML technologies, as the existing FDA medical device regulations were not specifically designed for them. Continuously learning AI/ML technologies have the potential to optimize device performance in real-time and continuously improve healthcare for patients.
The new framework would allow manufacturers to obtain pre-clearance or pre-approvals from the FDA for certain types of postmarket software changes without the need for new FDA submissions (e.g., new 510(k)s or new PMAs including PMA supplements, respectively). By setting up a PCCP with well-defined boundaries and implementing it as a part of the change control process under their existing quality management system (QMS), manufacturers could ensure the continued safety and effectiveness of the continuously learning AI/ML software. If implemented properly, this regulatory framework can unlock the full potential of continuously learning AI/ML technologies while providing reasonable assurance of software safety and effectiveness.
Since the publication of the 2019 FDA PCCP discussion paper, ongoing discussions have centered on what would be covered in the much-anticipated FDA draft guidance[2] titled “Marketing Submission Recommendations for A Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions,” and whether the FDA has the regulatory authority to implement such a program without additional congressional authorization.
In December 2022, the U.S. Congress passed the Consolidated Appropriations Act, 2023[3] (also known as the 2023 omnibus bill), which granted the FDA additional legal authority to implement the PCCP, per Section 3308 of the Food and Drug Omnibus Reform Act (FDORA). To the surprise of many who were not closely following the legislation process, the scope of the PCCP outlined in FDORA is broad, beyond AI/ML-enabled software, covering all medical devices requiring premarket submissions (i.e., premarket notifications or de novo classification requests for non-exempt Class II devices and premarket approvals (PMA) for the Class III devices). It is considered good news by industry, healthcare and academic communities, healthcare providers, and patients for the FDA to have clear regulatory authority to implement the PCCP for all devices requiring premarket submissions. This creates the opportunity to strike a better balance between promoting innovation and protecting public health.
The FDA recently published the draft guidance on April 3, 2023 – Draft Guidance for Industry and Food and Drug Administration Staff, Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions (also known as AI/ML PCCP draft guidance). As the guidance title suggests, this document only applies to AI/ML-enabled software functions.
Given the wide range of non-exempt Class II and Class III medical devices that fall under FDA regulation, each with its unique technological characteristics and safety profile, we expect that the FDA will implement the PCCP in phases, for example, addressing software before hardware and Class II before Class III. We anticipate that the FDA will likely codify the PCCP into regulations and/or issue multiple guidance documents which could be published in the following order of priority:
1. AI/ML-based Software as a Medical Device (SaMD) (As noted in Section 2.1, the FDA released the much-anticipated draft guidance on April 3, 2023.)
2. General Class II SaMDs subject to premarket notification.
3. General Class II medical devices subject to premarket notification.
4. Subsets of, or all, Class III devices subject to premarket approval.
The primary focus of this white paper is on general Class II SaMDs subject to premarket notification.
By publishing this white paper, we wish to provide a thought piece that can help accelerate the full rollout of effective PCCP regulations and guidance, and to achieve the following goals:
– Decipher the statutory requirements for the FDORA Section 3308 related to the PCCP.
– Provide considerations for selecting appropriate changes for the PCCP for Class II SaMD products.
– Propose key components of the PCCP for Class II SaMD products.
The discussions in this white paper will be limited to non-exempt Class II SaMD products, following a background on change controls for all medical devices and an interpretation of Section 3308 statutory language. While some of the principles and considerations may apply to other non-exempt Class II medical devices (e.g., traditional hardware devices with embedded firmware), we do not intend to fully address considerations for implementing the PCCP for non-exempt Software in a Medical Device (SiMD) functions given that the SiMD involves closer interactions with the hardware. For Class III PMA devices, the PCCP implementation strategy and considerations can be quite different and therefore outside the scope of this white paper.
To accomplish these objectives, to understand PCCP in the context of the general change controls practices and to incorporate PCCP into company’s existing change controls process, the main body of this white paper is structured as follows:
– Section 2.1 describes the general change controls process for all medical devices.
– Section 2.2 reviews the current FDA guidance regarding modifications for all 510(k) devices.
– Section 3 interprets the statutory language in Section 3308 of the FDORA related to PCCP, which applies to all Class II and Class III devices requiring premarket submissions.
– Section 4 provides considerations for selecting suitable changes for the 510(k) SaMD product PCCP to increase the chance of success.
– Section 5 proposes appropriate components for an effective PCCP for the 510(k) SaMD products.
– Section 6 addresses frequently asked questions and identifies other open questions that deserve further dialogue between the medical device industry and the FDA.
One of the key defining characteristics of medical devices is that the device manufacturers will continue to make changes after the initial commercialization of the medical devices. Such post-market changes can be planned or unplanned. They can be made for many reasons including:
– To improve the device by adding new features or enhancing usability.
– To maintain or improve safety, effectiveness, quality and/or reliability.
Examples of planned changes include, but are not limited to:
– Expanding indications for use to include pediatric user populations.
– Adding new product features.
– Modifying the software user interface for better usability.
– Adding a new supplier for a key component to improve supply chain management.
– Switching from manual to automatic production lines.
Examples of unplanned changes include, but are not limited to:
– Switching to a new raw material supplier due to sudden bankruptcy of the existing supplier.
– Implementing a security patch for a vulnerability newly identified by a third party.
– Updating SaMD for a major operating system (OS) upgrade affecting Bluetooth communications.
– Starting identical production at a new manufacturing location after a strong earthquake has incapacitated the existing manufacturing facility.
The change control process is a key part of any medical device quality management system (QMS). To properly evaluate and implement any change made after the initial commercialization of the medical device, the change control process typically follows the six steps below:
1. Understand the proposed change.
The device manufacturer needs to understand the scope and details of a proposed change, and the reason for making such change.
2. Conduct an impact assessment on the change.
The device manufacturer needs to conduct a thorough impact assessment to understand the potential consequences of the proposed change on device safety, effectiveness, quality and/or reliability, whether such change may potentially affect other hardware components or software modules in the same device or other connected external devices or systems, and any potential changes to the existing risk assessment.
Risk assessment of the change is one of the most critical parts of the impact assessment process. The manufacturer needs to review the existing risk management documents for the device, and ask the key question – whether the proposed change could potentially affect the existing risk assessment. There are several ways that a proposed change could potentially affect the existing risk assessment, including:
i. Changing the severity and/or probability of occurrence of any existing harm, negatively or positively.
ii. Introducing a new hazard or hazardous situation.
iii. Making an existing risk control measure less effective.
iv. Requiring a new risk control measure.
To evaluate multiple changes (e.g., a component change with manufacturing process changes for component assembly), the impact assessment needs to be repeated for each proposed change and then for the cumulative effect of the changes combined.
3. Perform appropriate verification and/or validation testing.
Based on the outcome of the impact assessment, the manufacturer needs to determine and perform an appropriate level of verification and/or validation testing, including bench testing, animal testing, usability testing, security testing, and human clinical study.
In addition to generating evidence for continued safety, effectiveness, device quality and/or reliability, the verification and/or validation testing results may prompt the manufacturer to update the change impact assessment, including risk assessment, implement additional risk control measures, or revise regulatory assessment conclusion as described in Step 4 below.
4. Conduct regulatory assessment and submit an FDA submission when appropriate.
For non-exempt Class II 510(k) devices and Class III PMA devices, the device manufacturer must determine whether a new FDA submission (including 510(k), or PMA including original PMA and various PMA supplements, excluding PMA Annual Report) is required to be submitted for the change based on the output of Step 2’s impact assessment and according to the relevant FDA regulations and guidance. In certain circumstances, the manufacturer could use the testing results from Step 3 to modify its FDA submission decision. In the case that a submission is required for the proposed change, the manufacturer will need to submit a new 510(k), PMA or PMA supplement and wait for FDA clearance or approval prior to implementing the change.
If the regulatory assessment determines that a submission is not required, the manufacturer can proceed with change implementation after completion of change controls activities and documentation per its internal QMS requirements. Such regulatory assessment outcome is often referred to as document-to-file or letter-to-file.
For PMA devices, the manufacturer may determine that the potential impact of a change falls between a submission and a document-to-file, allowing the manufacturer to implement the change first, then notify the FDA by including such change in the next PMA Annual Report.
5. Implement the change.
The manufacturer implements the proposed change according to its QMS requirements.
6. Monitor the postmarket performance.
Following the implementation of the change, the manufacturer must continuously monitor real-world performance of the modified device and take appropriate action (such as failure mode analysis, corrective and preventive actions (CAPA), additional risk control, field safety notification, recall, etc.), according to its own QMS requirements.
On October 25, 2017, the FDA issued two updated guidance documents on 510(k) device modifications:
– Guidance for Industry and Food and Drug Administration Staff, Deciding When to Submit a 510(k) for a Change to an Existing Device[4] (also known as “510(k) device modification guidance”)
– Guidance for Industry and Food and Drug Administration Staff, Deciding When to Submit a 510(k) for a Software Change to an Existing Device[5] (also known as “510(k) software modification guidance”)
As this white paper focuses on Class II SaMD products, this section provides a brief overview of the existing 510(k) software modification guidance, which was written before the introduction of the term “PCCP.” The guidance includes the following flowchart recommended for making regulatory assessment decisions on proposed software changes.
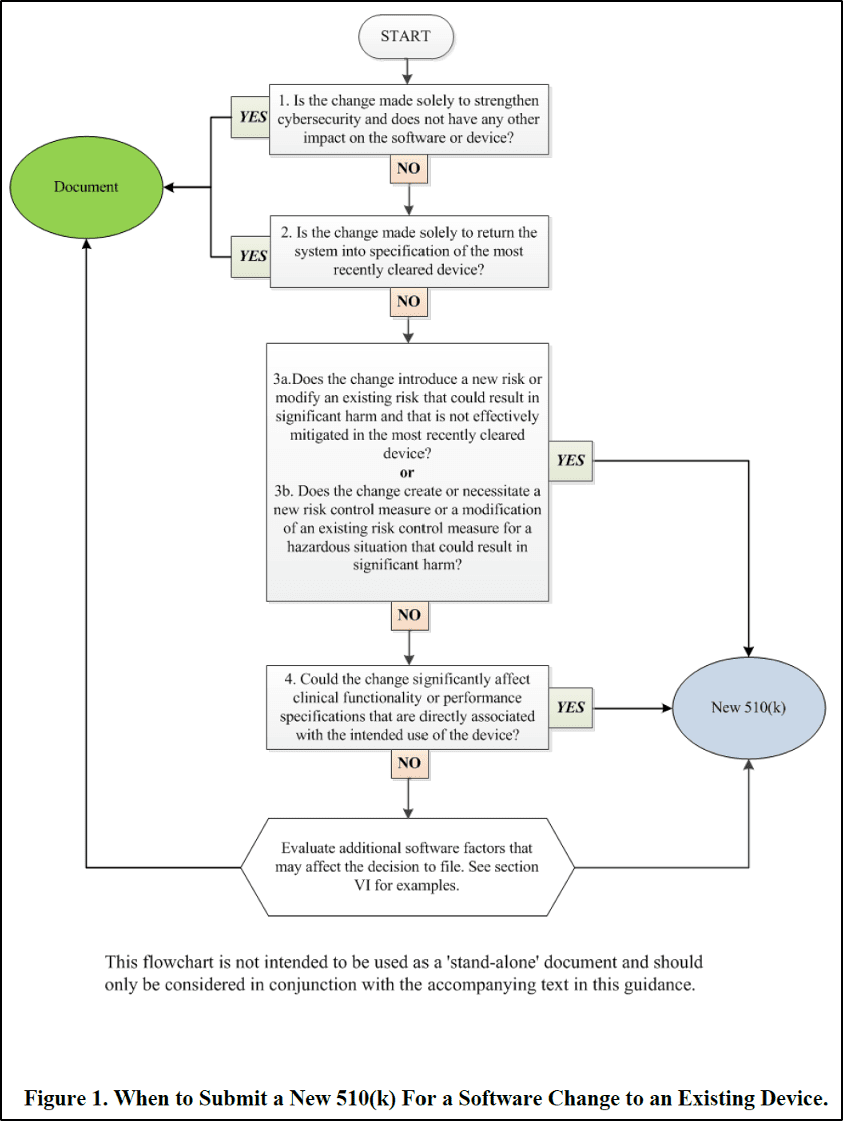
As illustrated in the flowchart and explained in detail in the accompanying text in the guidance, the questions in the flowchart are designed for the manufacturer to determine whether a 510(k) submission is required for the proposed software change. A 510(k) submission is required if one or both criteria is met:
– A major change or modification in the intended use of the device.
– A change or modification in the device that could significantly affect the safety or effectiveness of the device or result in a significant change or modification in design, material, chemical composition, energy source, or manufacturing process.
If, in addition to the proposed software change, there are other changes that may or may not be associated with the software change, or if the impact assessment concludes that the proposed software change may affect other aspects of the device (i.e., indications for use, labeling or device performance, etc.), the 510(k) device modification guidance should also be used to determine whether a new 510(k) needs to be submitted to the FDA.
If the manufacturer determines that, based on the regulatory assessment, a new 510(k) is not necessary, then the manufacturer can write a document-to-file and implement the proposed change upon successful completion of the required verification and/or validation testing.

As is typical with legislative language, the language used in Section 3308 of the Food and Drug Omnibus Reform Act (FDORA) can be difficult to understand, with multiple cross-references to existing laws and use of legal terminology that can be cryptic. Eventually, the FDA will clarify the new law by updating the existing code of federal regulations (CFR) and/or by issuing guidance documents.
In the meantime, we provide our initial interpretation of Section 3308 in the following table to assist medical device manufacturers in better understanding the new authority granted to the FDA regarding PCCP.
Section 3308 of the FDORA contains two subsections:
– The first subsection, 3308 (a), adds a new section to the Food, Drug and Cosmetic Act (FD&C Act) called the Predetermined Change Control Plans for Devices, which is designated as Section 515C.
– The second subsection, 3308 (b), makes “conforming amendments” to the existing sections of the FD&C Act, which ensures the newly added Section 515C works in concert with other existing sections without creating any conflicts.
SEC. 3308. PREDETERMINED CHANGE CONTROL PLANS FOR DEVICES.
Legislative Text: (a) IN GENERAL.—Chapter V of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 351 et seq.) is amended by inserting after section 515B (21 U.S.C. 360e–3) the following:
Interpretation: 3308(a) adds a new section to the Federal Food, Drug and Cosmetic Act (Federal FD&C Act) called the Predetermined Change Control Plans for Devices, designated as Section 515C.
SEC. 515C. PREDETERMINED CHANGE CONTROL PLANS FOR DEVICES.
Legislative Text: (a) APPROVED DEVICES.—
Interpretation: 515C(a) is for Class III PMA devices (or approved devices)
Note: Although the new law does not specifically reference humanitarian device exemption (HDE), it is important to note that HDE devices can also be approved under Section 515 of the Federal FD&C Act as Class III devices. It is likely that the FDA may allow PCCPs to be submitted under an original HDE or a HDE supplement submission. For the purpose of this white paper, we will use the terms “PMA devices” and “PMA submissions” to include HDE devices and HDE submissions hereafter, respectively.
Legislative Text: (1) IN GENERAL.—Notwithstanding section 515(d)(5)(A), a supplemental application shall not be required for a change to a device approved under section 515, if such change is consistent with a predetermined change control plan that is approved pursuant to paragraph (2).
Interpretation: (1): A PMA supplement is not required for a change made to a PMA device if such change is within the scope of and consistent with an authorized PCCP.
Legislative Text: (2) PREDETERMINED CHANGE CONTROL PLAN.—The Secretary may approve a predetermined change control plan submitted in an application, including a supplemental application, under section 515 that describes planned changes that may be made to the device (and that would otherwise require a supplemental application under section 515), if the device remains safe and effective without any change.
Interpretation: (2): The FDA may approve a PCCP submitted under an original PMA or a PMA supplement for planned or anticipated future changes to a PMA device. Such future changes may otherwise require PMA or PMA supplement submissions without being pre-authorized under a PCCP.
– We interpret the conditional statement, “if the device remains safe and effective without any change,” under (a)(2) refers to future changes implemented under the authorized PCCP should not include changes that are intended to fix a baseline device that has significant safety or effectiveness issues. For example, even if a proposed change is within the scope of the authorized PCCP, if such change is made to fix significant safety and/or effectiveness issues existing in the baseline device, the FDA will likely require a submission (e.g., change being effected or CBE, or a PMA supplement).
Legislative Text: (3) SCOPE.—The Secretary may require that a change control plan include labeling required for safe and effective use of the device as such device changes pursuant to such plan, notification requirements if the device does not function as intended pursuant to such plan, and performance requirements for changes made under the plan.
Interpretation: (3): The FDA may require the PCCP to include certain elements, such as product labeling to ensure continued safety and effective use of the modified device by the intended users, performance criteria for changes implemented under the PCCP, and control plan, such as customer notification if implemented change does not perform in the field as intended. Depending on the nature and degree of the performance issue, a correction or removal report to the FDA and/or a product recall may be necessary under certain circumstances.
Legislative Text: (b) CLEARED DEVICES.—
Interpretation: 515C(b) is for non-exempt Class II 510(k) devices (i.e., cleared devices).
Note: “Cleared devices” include non-exempt Class II devices granted through de novo classification requests.
Legislative Text: (1) IN GENERAL.—Notwithstanding section 510(k), a premarket notification shall not be required for a change to a device cleared under section 510(k), if such change is consistent with an established predetermined change control plan granted pursuant to paragraph (2).
Interpretation: (1): A new 510(k) is not required for a change made to a 510(k) device if such change is within the scope of and consistent with an approved PCCP.
Legislative Text: (2) PREDETERMINED CHANGE CONTROL PLAN.—The Secretary may clear a predetermined change control plan submitted in a notification submitted under section 510(k) that describes planned changes that may be made to the device (and that would otherwise require a new notification), if—
‘‘(A) the device remains safe and effective without any such change; and
‘‘(B) the device would remain substantially equivalent to the predicate.
Interpretation: (2): The FDA may approve a PCCP submitted under a 510(k) or a de novo for planned or anticipated future changes to a non-exempt Class II 510(k) device. Such future changes may otherwise require new 510(k) submissions without being pre-cleared under a PCCP, per the criteria explained in Section 2.2.
– We expect that a PCCP can be submitted in a de novo classification request for a new non-exempt Class II regulation. Once the de novo request is granted, such device becomes a non-exempt Class II 510(k) device.
– We interpret that the two conditional statements, “if (A) the device remains safe and effective without any such change; and (B) the device would remain substantially equivalent to the predicate,” under (b)(2) refer to future changes implemented under the authorized PCCP should not include changes that either (A) are intended to fix a baseline device that has significant safety or effectiveness issues, or (B) make the modified devices no longer substantially equivalent to the last cleared devices. These are the exceptions to implementing changes per PCCP without a new 510(k) submission.
– If a change is made to fix significant safety and/or effectiveness issues existing in the baseline device, the FDA will likely require a submission (e.g., a CBE 510(k) or a 510(k)) for such a change that is otherwise consistent with the PCCP. If the modified device with a proposed future change will no longer be substantially equivalent to the last cleared device (i.e., the predicate device), the FDA will likely require a submission – a de novo or a 510(k) – for such change.
Legislative Text: (3) SCOPE.—The Secretary may require that a change control plan include labeling required for safe and effective use of the device as such device changes pursuant to such plan, notification requirements if the device does not function as intended pursuant to such plan, and performance requirements for changes made under the plan.
Interpretation: (3): The FDA may require the PCCP to include certain elements, such as product labeling to ensure continued safety and effective use of the modified device by the intended users, performance criteria for changes implemented under the PCCP, and control plan, such as customer notification if implemented change does not perform in the field as intended. Depending on the nature and degree of the performance issue, a correction or removal report to the FDA and/or a product recall may be necessary under certain circumstances.
Legislative Text: (c) PREDICATE DEVICES.—In making a determination of substantial equivalence pursuant to section 513(i), the Secretary shall not compare a device to changed versions of a device implemented in accordance with an established predetermined change control plan as a predicate device. Only the version of the device cleared or approved, prior to changes made under the predetermined change control plan, may be used by a sponsor as a predicate device.’’
Interpretation: (c): Changed versions of a 510(k) device implemented under an authorized PCCP should not be used as the predicate device for future 510(k) premarket notifications. All future 510(k)s submitted by the same manufacturer for any change that (1) is not consistent with the PCCP and therefore not pre-authorized, AND (2) would trigger a new 510(k) submission, or 510(k)s submitted by different manufacturers, should use a predicate device, either a device version cleared by the FDA through a 510(k) premarket notification or granted through a de novo classification request.
Legislative Text: (b) CONFORMING AMENDMENTS.—
Interpretation: 3308 (b) makes conforming amendments to the existing sections of the Federal FD&C Act, which ensures the newly added Section 515C works in concert with other existing sections without creating any conflicts.
Legislative Text: (1) CLEARED DEVICES.—Section 510(l)(1) of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 360(l)(1)) is amended, in the first sentence, by inserting ‘‘, or with respect to a change that is consistent with a predetermined change control plan cleared under section 515C’’ before the period at the end.
Interpretation: (1): Adding “or with respect to a change that is consistent with a predetermined change control plan cleared under section 515C” to 21 U.S.C. 360(l)(1) (or Federal FD&C Act, Section 510(l)(1)) for non-exempt Class II 510(k) devices.
With the edits shown in the following paragraph with underlines for new texts, the amended 21 U.S.C. 360(l)(1) now reads the following (Note: “Report under subsection (k)” refers to 21 U.S.C. 360(k) or Federal FD&C Act 510(k) premarket notification requirement):
§360. Registration of producers of drugs or devices
(l) Exemption from reporting requirements
(1) A report under subsection (k) of this section is not required for a device intended for human use that is exempted from the requirements of this subsection under subsection (m) of this section or is within a type that has been classified into class I under section 360c of this title, or with respect to a change that is consistent with a predetermined change control plan cleared under section 515C. The exception established in the preceding sentence does not apply to any class I device that is intended for a use which is of substantial importance in preventing impairment of human health, or to any class I device that presents a potential unreasonable risk of illness or injury.
Legislative Text: (2) APPROVED DEVICES.—Section 515(d)(5)(A)(i) of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 360e(d)(5)(A)(i)) is amended by striking ‘‘A supplemental’’ and inserting ‘‘Unless the change is consistent with a predetermined change control plan approved under section 515C, a supplemental.”
Interpretation: (2): Removing ‘‘A supplemental’’ and adding ‘‘Unless the change is consistent with a predetermined change control plan approved under section 515C, a supplemental’’ to 21 U.S.C. 360e(d)(5)(A)(i) (or Federal FD&C Act, Section 515(d)(5)(A)(i)) for Class III PMA devices.
With the edits shown in the following paragraph with strikethroughs for removed texts and underlines for new texts, the amended 21 U.S.C 360e(d)(5)(A)(i) now reads the following:
§360e. Premarket approval
(d) Action on application for premarket approval
(5)(A)(i) A supplemental Unless the change is consistent with a predetermined change control plan approved under section 515C, a supplemental application shall be required for any change to a device subject to an approved application under this subsection that affects safety or effectiveness, unless such change is a modification in a manufacturing procedure or method of manufacturing, and the holder of the approved application submits a written notice to the Secretary that describes in detail the change, summarizes the data or information supporting the change, and informs the Secretary that the change has been made under the requirements of section 360j(f) of this title.
Legislative Text: (3) DOCUMENTATION OF RATIONALE FOR SIGNIFICANT DECISIONS.—Section 517A(a)(1) of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 360g–1(a)(1)) is amended to read as follows:
‘‘(1) IN GENERAL.—The Secretary shall provide a substantive summary of the scientific and regulatory rationale for any significant decision of the Center for Devices and Radiological Health regarding submission or review of a report under section 510(k), a petition for classification under section 513(f), an application under section 515, or an application for an exemption under section 520(g), including documentation of significant controversies or differences of opinion and the resolution of such controversies or differences of opinion.’’
Interpretation: (3): Amending Section 517A(a)(1) of the Federal FD&C Act.
With the edits shown in the following paragraph with strikethroughs for removed texts and underlines for new texts, the amended Federal FD&C Act, Chapter V, Section 517A(a)(1) now reads the following (Note: Section 517A pertains to requests for appeals of “significant decisions” concerning submissions under sections 510(k) (premarket notification), 513(f) (de novo classification request or de novo), 515 (Premarket Approval or PMA/Humanitarian Device Exemption or HDE), or 520(g) (Investigational Device Exemption or IDE), of the Federal FD&C Act. Although the edits here are not directly related to the PCCP, since a PCCP can be submitted under a 510(k), a de novo, or a PMA or PMA supplement, and possibly under a HDE or HDE supplement, a manufacturer’s request for appeals of FDA’s decisions on these submissions could indirectly relate to the PCCPs submitted under such submissions.)
§517A AGENCY DOCUMENTATION AND REVIEW OF SIGNIFICANT DECISIONS REGARDING DEVICES
(a) DOCUMENTATION OF RATIONALE FOR SIGNIFICANT DECISIONS.
(1) IN GENERAL.—The Secretary shall provide a substantive summary of the scientific and regulatory rationale for any significant decision of the Center for Devices and Radiological Health regarding submission or review of a report under section 510(k), a petition for classification under section 513(f), an application under section 515, a request for designation under section 515B2 , or an application for an exemption under section 520(g), including documentation of significant controversies or differences of opinion and the resolution of such controversies or differences of opinion.
What has changed with the new statutory requirements under Section 3308 Predetermined Change Plans for Devices of the FDORA:
– The FDA now has clear statutory authority to exempt certain future changes to 510(k) devices and PMA devices from premarket notifications and premarket approval submissions, respectively, if such changes are consistent with the PCCP authorized by the FDA in a previous 510(k) (or de novo) or PMA submission.
What has not changed with the implementation of the PCCP under Section 3308:
– The premarket clearance and the premarket approval process, as well as their requirements, remain the same for all submissions (i.e., 510(k), de novo, and PMA).
– For any premarket notification 510(k) submission, the eligible predicate device requirement remains the same, i.e., needs to be a device version that is cleared through the 510(k) process or granted through the de novo classification process.
As interpreted in Section 3.1 of this white paper, the new section 515C(c) clarifies that any device version implemented according to a PCCP cannot be used as a predicate device for any future 510(k) submission.
– – – – –
This blog contains Part 1 of the Orthogonal and MedSec white paper titled “Making the Significant Insignificant: Implementing a Predetermined Change Control Plan (PCCP) for Class II SaMD Products Beyond AI/ML.” The following are links to each part of this white paper:
White Paper

Webinar

White Paper

Webinar

Article

White Paper

Article

Webinar
