
Article
Why Ecosystem Design Controls Are Really About Moving Faster With the Right Rigor


Orthogonal is pleased to publish this guest blog by Don Peters, president of dPeters Consulting. Don has thirty six years industrial experience with product development in life sciences and medical devices. He has contributed to or led teams in software/systems engineering resulting in multiple successful product launches ranging from clinical diagnostics to ophthalmic surgical equipment. Don created dPeters Consulting, LLC in 2016 and has consulted with multiple clients supporting technologies in life sciences, pharma and medical devices. Don specializes in quality management systems related to the software development life cycle and risk management with a focus on the client’s organizational interfaces between Systems Development, Quality Assurance and Regulatory Affairs.
Don Peters is an accomplished and impressive “friend of Orthogonal.” He shares our belief that the MedTech industry can improve patient outcomes faster by adopting fast feedback loop techniques to the development and enhancement of Software as a Medical Device (SaMD), digital therapeutics (DTx) and connected medical device systems.
Don writes about one of the thorniest problems in our industry: how do we define the process of validation? Don offers practical advice and steps to take to overcome a lack of clarity or understanding in what is a new, subtle and uncharted territory.
You can reach Don at [email protected]
The term GxP includes clinical (GCP), manufacturing (GMP), distribution (GDP), laboratory (GLP), etc. Each GxP organization establishes trust with their customers (internal or external) through validation. Validation is especially important when software is involved. For the purpose of this article, “System” will refer to systems containing software that are embedded in hardware or virtualized elsewhere. Systems are represented in products, services, tools, quality systems and manufacturing systems.
Simply put, validation is a risk-based approach to demonstrate a given system is fit for its intended use throughout the System’s life-cycle.[1-11] This applies broadly to products, services, production and quality systems for all GxP organizations.
System validation is what makes a regulated GxP organization’s software whole. And yet, the above simple phrase seems to flummox even those seasoned in the medical device industry and systems containing software. There is a lack of clarity of what the standards are saying; the nomenclature is not being interpreted correctly. Most system developers are very pragmatic and detailed in their design, implementation, evaluation, deployment and maintenance documentation, but even they are confused on what to do when it comes to validation.
In my experience, these problems exist in many GxP organizations, medical device and non-medical device companies, regardless of size or product complexity. The following concerning observations in Table 1 were found by regulators during FDA Inspections in 2020 and 2021 with regards to 21 CFR 820: [12-14]

A concerning gap listed above is the lack of application or documentation of risk analysis. From my perspective working in the GxP industry for several years, these are not new observations. Rather, these are systemic issues rooted in a lack of understanding of a handful of fundamental principles. These observations contain common elements that support the simple validation phrase above; regardless of medical device or non-medical device industries.
Emerging technologies in software have accelerated over the last couple of years including: Data Analytics; Cloud computing; IoT; Interoperability; Mobile Devices; and AI/ML. The adoption of these technologies will widely impact product development, manufacturing, deployment and support as well as software systems supporting the entire enterprise infrastructure. These technologies challenge the traditional approach to validation and will likely require more frequent, rapid and widely deployed changes.[15-19]
Concurrently, concerns increase for GxP “critical infrastructure” (e.g. manufacturing plants) regarding accessibility, cybersecurity, data integrity, data security as well as human safety.[20-22] For example, regulated GxP organizations are beginning to recognize the traditional enterprise security perimeters may no longer exist.[23] Most technologies are likely to be directly exposed to the internet, putting systems at significantly greater risk of compromise. Furthermore, many GxP organizations are interested in utilizing emerging technologies to develop products involving: digital health; clinical decision software; interoperability with clinical systems; health informatics and of course – Software as a Medical Device (SaMD)[24] and digital therapeutics. The adoption of these new technologies will require increased attention to a system’s risk management file over the entirety of its lifecycle.
One of the challenges of System Validation, including medical device software, is that there is no single standard that consistently explains software validation. Rather, validation has a subtle palette of different flavors that needs to be applied properly to be effective; especially when software is involved. [1,2,5,10,11] The standards use various nuances and terms for system validation involving: Products & Services; Automated Manufacturing Equipment; Quality System applications; other systems used in Production & Services Provision; Monitoring and Measurement Systems.
Perhaps clarification is needed to describe these flavors and how they apply to fundamental elements of validation. The standards and guidance documents listed in Table 2 (and additional citations) are used to support a handful of useful Validation Tips. These tips are generalized for all GxP organizations based on these standards and guidance. Note, it may be interesting that many of the concepts found in ISO 13485 and FDA 21 CFR 820, were originally derived from ISO 9001.[25]
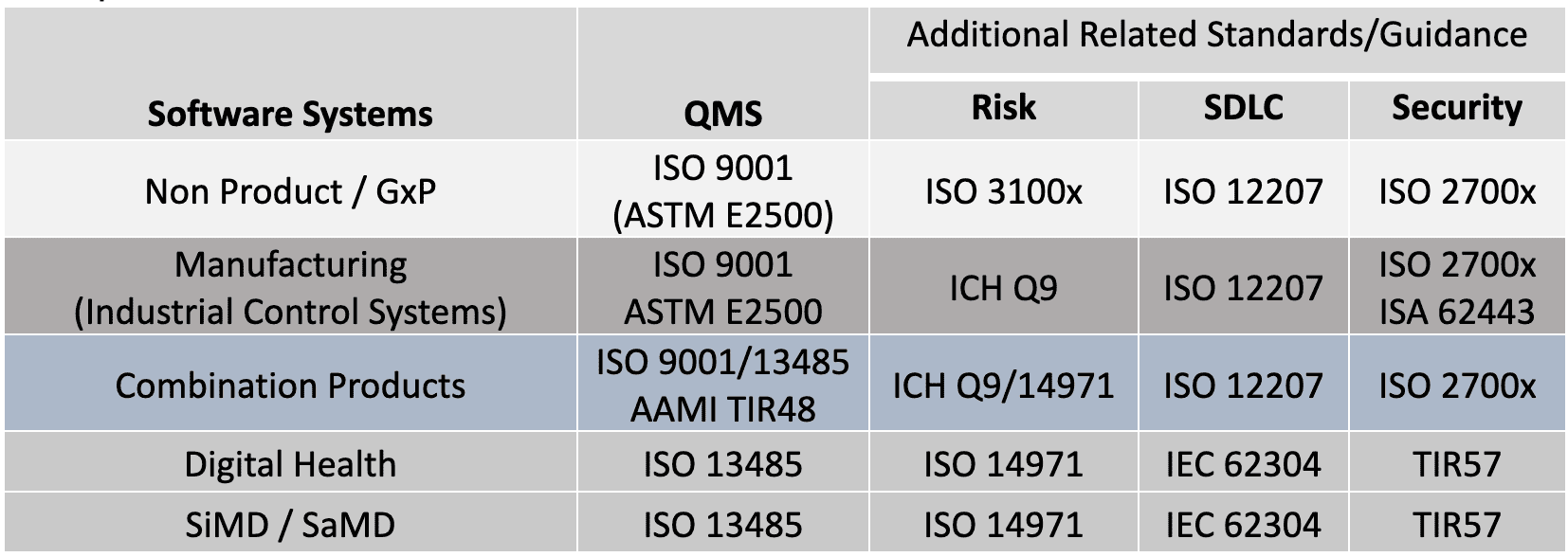
Table 2: Quality System Regulations, Standards and Guidance Across GxP
There are common structures and processes to these standards in Table 2. In my analysis, I’ve found a common set of overlapping elements that can be used to better understand the approach to validation. The following tips can be applied to: new systems development; maintenance of existing systems; systems that support the enterprise including critical QMS applications or manufacturing systems.
Before starting any System validation activity, it is important to recognize the System’s state within its lifecycle[11] (see Figure 1). Although it may get the most attention in the industry, the original development process through deployment might represent just 10-20% of the overall lifecycle activities devoted to validation. However, maintenance activities will likely take the majority of the remaining lifecycle including re-validation efforts. As implied with the above FDA observations, maintenance activities may be compromised without a robust initial system baseline. At a minimum, the risk management file (baseline) needs to be reviewed for any change impacts.[2,7,11,26] Certain design changes including security changes may require an updated risk file. Revalidation efforts must ensure the system remains in a validated state.[5, 8, 11,16, 27]
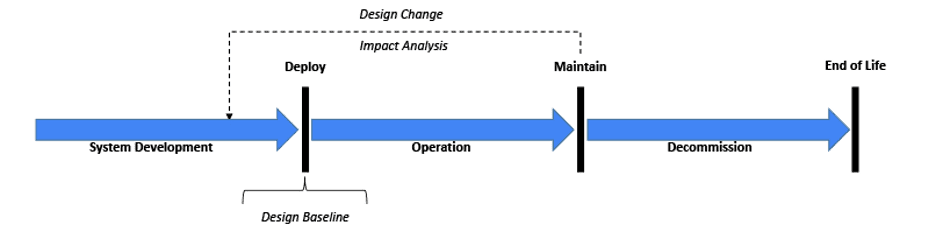
Figure 1: Generalized System Lifecycle
Next, let’s look at the relationship between: Intended Use; Risks and Requirements (Figure 2). The intended use[7,10] (or purpose) needs to be taken in context of the environment it is used (or acted on) and the stakeholders that may be involved.[2, 5, 11, 27, 29, 30] This intended use set is important to establish risks based on reasonably foreseeable misuse.[31] An organization with a low risk profile may only need to establish Control Measures for quality attributes (e.g. functions, features, abilities, performance, characteristics)[10] that are critical to Satisfaction and Performance. Other organization’s may have a higher risk profile and will need to go deeper.[7, 10, 32, 33]
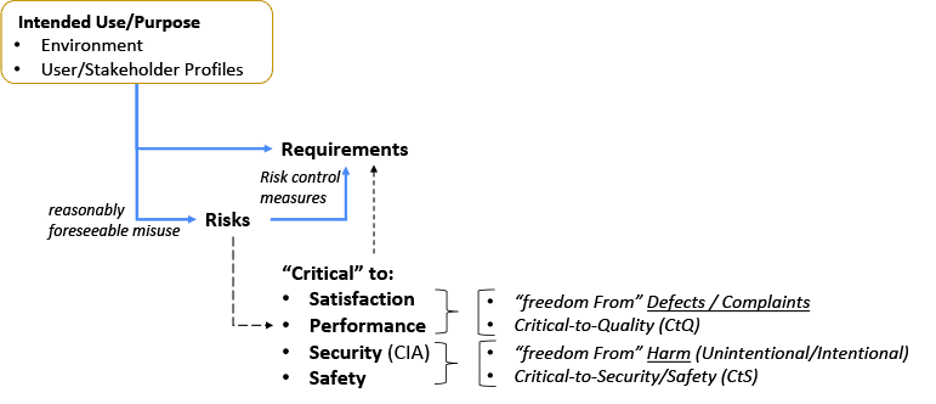
Figure 2: Intended Use Relationship with Risks and Requirements
The illustration in Figure 3 considers the relationship between the Risk Management File (RMF) and the System design. The Risk Management diagram is a simplified illustration common to multiple standards including.[26,32,35-38] The terms within the RMF illustration are unique and important. (However, terminology may differ, such as: control measures; countermeasures; risk mitigation; or risk treatment.) The intended use and various other design elements are fed into the Risk Assessment as input. It is noteworthy that the Intended Use and Customer Needs may overlap, but are not necessarily the same thing.[8, 27, 30] Risk control measures need to be linked or written into requirements, and verified.[8, 27]
A V-diagram is used to illustrate traceability across content and indicate the depth of design details. This diagram is not intended to imply any development framework (i.e. waterfall, agile, etc). This diagram depth may be shallower or deeper depending on the complexity of the system. But the design depth may also be constrained to its Degree of Design Ownership, or the control one has over the system design.
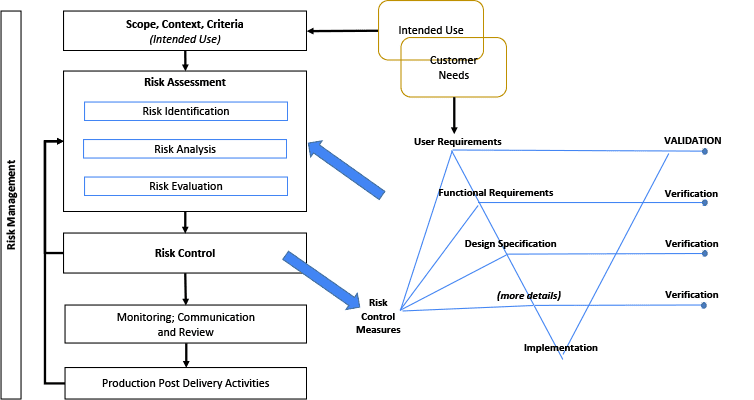
Figure 3: Relationship Between Risk Management File and the System Design
Selecting an appropriate approach to validate a given system depends on several factors including the intended use, users and environment the system is expected to function and perform. But the validation approach also depends on the degree of ownership (or control) one has over the system to be validated. A manufacturer that develops a medical device typically owns and maintains the system requirements, design specification and implementation where there is a high degree of ownership over the system. Whereas the same manufacturer may acquire software applications (e.g. Off the Shelf (OTS)) that support the development process (e.g. automated testing or defect tracking). Or the same manufacturer may acquire software to support manufacturing processes or quality system applications to support a product in the market (e.g. Complaint tracking). These applications may not include significant design documentation other than a basic design specification summary, information for use. But these acquired applications may require significant modification or configuration to satisfy a particular workflow or process.
The methods used to validate a given system depends on: the complexity of the design; the intended use – including the users and users environment; and the risks associated with its use. This is typically known as Design Validation. But what if the design details are not available as described above? There are caveat provisions cited in the standards that address this situation:
Under these circumstances, Process Validation applies to automated software systems[1-2,4,9-10,26] and includes:
Objective evidence[2,5,9-10,26] showing the confirmation[1,2,5,9-10] the system’s requirements[2,5,26] can consistently fulfill[1-2,4,5] its intended use.[2,4-5,10] A risk based[4,,9,10,26] (including safety & critical attributes of quality)[9-10] is used to determine the scope and extent of the verification and validation/revalidation[9-10,26] activities, throughout the systems life cycle.[4,9,10]
Some terms used in Process Validation are different from what’s used in Design Validation.[2] The word validation is sometimes widened to incorporate the concept of qualification.[9-10] Qualification includes verification of critical aspects (product quality and patient safety), Critical Process Parameters; and risk control measures.[10] And, verification is sometimes called a qualification process.[30]
Two examples are illustrated in Figure 4 to indicate either end of a spectrum for design ownership. Two V-diagrams are used to demonstrate the presence or absence of design documentation depth.
An organization wanting to use acquired components or applications is an example for the Low Design Ownership diagram. Design documentation may be limited or not available from a supplier. The organization using these components or applications need to create and overlay their own documentation to define their intended use, risk, functional requirements, configurations, etc.[11] Verification may be limited to only the organizations overlaying documentation to confirm overlaying risk control measures, functional requirements and design specification. Validation takes all of this into consideration along with confirming the System’s fitness for intended use.
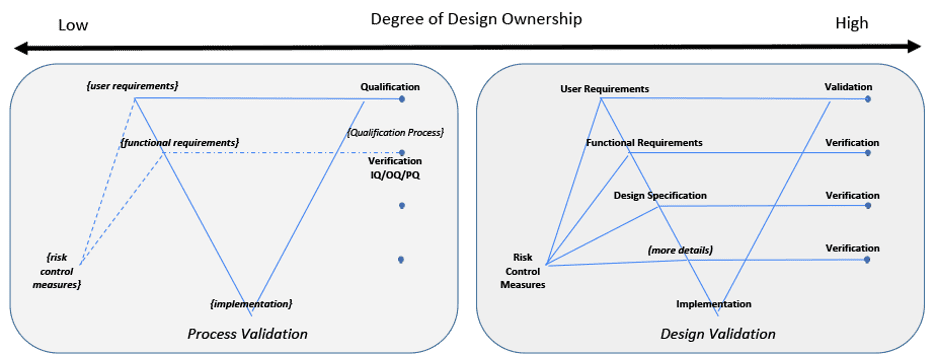
Figure 4: Degree of Design Ownership
The activities used in software validation are intended to establish a level of confidence that a system is appropriate for intended use and is trustworthy, reliable; and ensures software meets its requirements and intended purpose.[2,7,11,30]
Emerging technologies will provide GxP organizations new opportunities for improving the enterprise infrastructure, operations and product development. However, these opportunities come with additional potential risks in security and safety. Recent observations during regulatory inspections for medical devices may be a bellwether for other GxP industries regarding the understanding and appropriate use of validation. Extracting common and key elements specific to validation from either ISO 9001 or ISO 13485 related standards can provide simple constructs and processes that can be applied to any GxP organization. Whether your team is engaged in the initial development lifecycle stages (pre-delivery) or post-delivery maintenance activities, this article provides useful tips for defining a common language and methods to plan for a more effective and efficient validation effort. These tips can be used today to establish a high-level validation checklist, and can be used tomorrow to enhance your system development lifecycle procedures.
Citations 1. Guidance for Industry Process Validation: General Principles and Practices. Fda.gov. https://www.fda.gov/files/drugs/published/Process-Validation--General-Principles-and-Practices.pdf. Published 2011. Accessed March 17, 2022. 2. General Principles of Software Validation; Final Guidance for Industry and FDA Staff. Fda.gov. https://www.fda.gov/media/73141/download. Published 2002. Accessed March 17, 2022. 3. Blood Establishment Computer System Validation in the User’s Facility. Fda.gov. https://www.fda.gov/files/vaccines%2C%20blood%20%26%20biologics/published/Guidance-for-Industry--Blood-Establishment-Computer-System-Validation-in-the-User%27s-Facility.pdf. Published 2013. Accessed March 17, 2022. 4. E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1). Fda.gov. https://www.fda.gov/media/93884/download. Published 2018. Accessed March 17, 2022. 5. CFR - Code of Federal Regulations Title 21. Accessdata.fda.gov. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=820. Published 2022. Accessed March 17, 2022 6. ANSI/AAMI/IEC 62304:2006 & A1:2016. Store.aami.org. https://store.aami.org/s/store#/store/browse/detail/a152E000006j5rjQAA. Published 2015. Accessed March 17, 2022. 7. ISO/IEC/IEEE 12207:2017. ISO. https://www.iso.org/standard/63712.html. Published 2017. Accessed March 17, 2022. 8. ISO 13485:2016. ISO. https://www.iso.org/standard/59752.html. Published 2016. Accessed March 17, 2022. 9. EudraLex - Volume 4. Public Health. https://europa.eu/!9YmPyt. Published 2022. Accessed March 17, 2022. 10. Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment. Astm.org. https://www.astm.org/e2500-20.html. Published 2020. Accessed March 17, 2022. 11. ISO/TR 80002-2:2017. ISO. https://committee.iso.org/standard/60044.html. Published 2017. Accessed March 17, 2022. 12. The Top 10 Most-Cited Clauses In FDA FY2021 Medical Device Inspections. Meddeviceonline.com. https://www.meddeviceonline.com/doc/the-top-10-most-cited-clauses-in-fda-fy2021-medical-device-inspections-0001. Published 2022. Accessed March 17, 2022. 13. The Top 10 Most-Cited Issues In FDA FY2020 Medical Device Inspections. Meddeviceonline.com. https://www.meddeviceonline.com/doc/the-top-most-cited-issues-in-fda-fy-medical-device-inspections-0001. Published 2021. Accessed March 17, 2022. 14. Inspection Observations. U.S. Food and Drug Administration. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations. Published 2020. Accessed March 17, 2022. 15. AAMI CR510:2021 - Appropriate Use of Public Cloud Computing for Quality Systems and Medical Devices. Webstore.ansi.org. https://webstore.ansi.org/Standards/AAMI/AAMICR5102021. Published 2021. Accessed March 17, 2022. 16. GAMP RDI Good Practice Guide: Data Integrity by Design. ISPE | International Society for Pharmaceutical Engineering. https://ispe.org/publications/guidance-documents/gamp-rdi-good-practice-guide-data-integrity-design. Published 2020. Accessed March 17, 2022. 17. Anselmo C, Attili M, Horton R, Kappe B, Schulman J, Baird P. Hey You, Get On the Cloud: Safe and Compliant Use of Cloud Computing with Medical Devices. AAMI BI&T. https://meridian.allenpress.com/bit/article/55/1/1/451464/Hey-You-Get-On-the-Cloud-Safe-and-Compliant-Use-of. Published 2021. Accessed March 17, 2022. 18. Warner, Ph.D K. AI & Intelligent Technologies: Finding The Right Fit For Your Pharma Or Medtech Company. Outsourcedpharma.com. https://www.outsourcedpharma.com/doc/ai-intelligent-technologies-finding-the-right-fit-for-your-pharma-or-medtech-company-0001. Published 2022. Accessed March 17, 2022. 19. Miller M, Zaccheddu N. Light for a Potentially Cloudy Situation: Approach to Validating Cloud Computing Tools. AAMI BI&T. https://meridian.allenpress.com/bit/article-abstract/55/2/63/465469/Light-for-a-Potentially-Cloudy-Situation-Approach?redirectedFrom=fulltext. Published 2021. Accessed March 17, 2022. 20. Cisa.gov. https://www.cisa.gov/sites/default/files/publications/dhs-eo13636-analytic-report-cybersecurity-incentives-study.pdf. Published 2013. Accessed March 17, 2022. 21. Executive Order 14028, Improving the Nation's Cybersecurity. NIST. https://www.nist.gov/itl/executive-order-improving-nations-cybersecurity. Published 2022. Accessed March 17, 2022. 22. Stouffer K, Pillitteri V, Lightman S, Abrams M, Hahn A. Guide to Industrial Control Systems (ICS) Security. NIST. https://csrc.nist.gov/publications/detail/sp/800-82/rev-2/final. Published 2015. Accessed March 17, 2022. 23. Rose S, Borchert O, Mitchell S, Connelly S. Zero Trust Architecture. NIST. https://www.nist.gov/publications/zero-trust-architecture. Published 2020. Accessed March 17, 2022. 24. Eggers W, Kishnani P, Turley M. The future of regulation. Deloitte Insights. https://www2.deloitte.com/xe/en/insights/industry/public-sector/future-of-regulation/regulating-emerging-technology.html. Published 2018. Accessed March 17, 2022 25. Current Good Manufacturing Practice Final Rule; Quality System. U.S. Food and Drug Administration. https://www.fda.gov/medical-devices/quality-system-qs-regulationmedical-device-good-manufacturing-practices/medical-devices-current-good-manufacturing-practice-cgmp-final-rule-quality-system-regulation 26. Q9 Quality Risk Management. Fda.gov. https://www.fda.gov/media/71543/download. Published 2006. Accessed March 17, 2022. 27. ISO 9001:2015. ISO. https://www.iso.org/standard/62085.html. Published 2015. Accessed March 17, 2022. 28. Medicines & Healthcare products Regulatory Agency (MHRA). Assets.publishing.service.gov.uk. https://assets.publishing.service.gov.uk/government/MHRA_GxP_data_integrity_guide. Published 2018. Accessed March 17, 2022. 29. ISO/IEC Guide 63:2019. ISO. https://www.iso.org/standard/67944.html. Published 2019. Accessed March 17, 2022. 30. ISO 9000:2015. ISO. https://www.iso.org/standard/45481.html. Published 2015. Accessed March 17, 2022. 31. ISO/IEC Guide 51:2014. ISO. https://www.iso.org/standard/53940.html. Published 2014. Accessed March 17, 2022. 32. ISO 14971:2019. ISO. https://www.iso.org/standard/72704.html. Published 2019. Accessed March 17, 2022. 33. IEC 80001-1:2010. ISO. https://www.iso.org/standard/44863.html. Published 2010. Accessed March 17, 2022. 34. ISO 31000 — Risk management. ISO. https://www.iso.org/iso-31000-risk-management.html/. Accessed March 17, 2022. 35. IEC 31010:2019. ISO. https://www.iso.org/standard/72140.html. Published 2019. Accessed March 17, 2022. 36. ISO/IEC 27001 Information security management. ISO. https://www.iso.org/isoiec-27001-information-security.html. Accessed March 17, 2022. 37. ISO/IEC 27005:2018 Information security risk management. ISO. https://www.iso.org/standard/75281.html. Published 2018. Accessed March 17, 2022. 38. GAMP Good Practice Guide: Enabling Innovation. ISPE | International Society for Pharmaceutical Engineering. https://ispe.org/publications/guidance-documents/gamp-good-practice-guide-enabling-innovation. Published 2021. Accessed March 17, 2022.
Related Posts
Article
Why Ecosystem Design Controls Are Really About Moving Faster With the Right Rigor

Article
MedTech Teams Should Stop Paying for Compliance Twice

Article
Beyond the Device: Where MedTech Digital Ecosystems Are Starting to Pay Off

Article
Why MedTech Teams Slow Down as Software Grows and What Better Architecture Looks Like
